Dr. Reddy’s Lab tanked on Friday and then again today, after news came out that involved US action against the company.
The stock has been in news for 2 different reasons:
- On Friday, USFDA (Food & Drug Administration) issued a warning letter in relation to the for the inspection performed API manufacturing facilities at Srikakulam, Andhra Pradesh and Miryalaguda, Telangana, as well as the Oncology Formulation manufacturing facility at Duvvada, Visakhapatnam, Andhra Pradesh. This is following the issuance of Form 483 containing the observations made during the earlier site inspections and the subsequent remediation plans submitted by Dr. Reddy’s.
- On Tuesday, the District Court of Delaware, USA granted in favour of Astra Zenecaand issued a Temporary Restraining Order for the sale, delivery, transfer or other disposition of its generic esomeprazole capsules (which reduces stomach acid secretion) in the US market. AstraZeneca happens to be the manufacturer of Nexium which is similar to Dr. Reddy’s esomeprazole capsules.
Let’s take a look at the stock as such:
[level-capmind-pro]
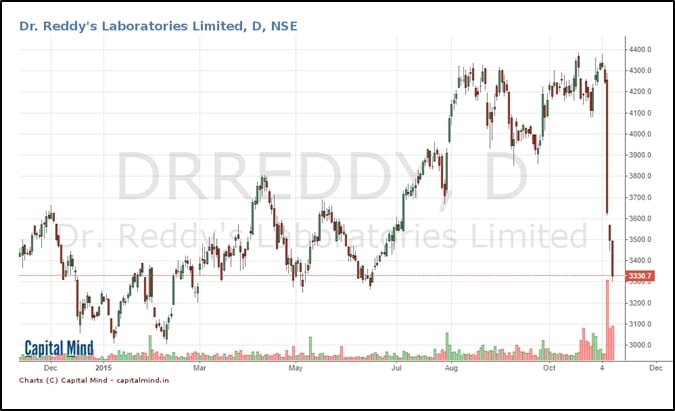
The stock has fallen back to the point where it took off in June 2015, and is about 10% higher than it’s 52 week lows. The impact has largely come in the last three days.
Now let’s get into the details of what happened : Part 1 – The US FDA site inspections
Week of 16 – 22 November, 2014 (One Year Back):
The Srikakulam facility in Andhra Pradesh region of India was inspected by the US Food and Drug Administration (FDA) during the week of 16th November.
Why did this take place? – All drugs approved in the United States, regardless of where they are made, must be in compliance with the Federal Food, Drug, and Cosmetic Act, which requires that drugs meet manufacturing standards to assure quality and product label requirements.
The USFDA typically inspects facilities, for pre-approval, compliance with current good manufacturing practices, or product specific approval. The FDA’s inspection team in India has around 20 people, who carry out ‘aggressive surveillance and surprise inspections. Since Dr.Reddy’s manufacturers most of its drugs in India, bulk of its products are exported.
Week of 23 – 29 November, 2014:
The inspection of the Srikakulam facility in Andhra Pradesh region resulted in the issuance of a Form 483 with nine observations. The observations were related to procedural and othercompliances of the plant system. It was an indication of non-confirmation to the manufacturing standards laid down by the regulatory authority.
This resulted in Dr. Reddy’s Srikakulam facility joining the USFDA’s list of offenders in 2014. The plant manufactures bulk active pharmaceuticals ingredients (APIs) – including bromfenacna, celecoxib, ibuprofen and others used in Dr. Reddy’s finished formulated drugs for export to the US and the EU.
What is an FDA Form 483? – An FDA Form 483 is issued to firm management at the conclusion of an inspection when an investigator(s) has observed any conditions that in their judgement may constitute violations of the Food Drug and Cosmetic (FD&C) Act and related Acts. FDA investigators are trained to ensure that each observation noted on the FDA Form 483 is clear, specific and significant. Observations are made when in the investigator’s judgement, conditions or practices observed would indicate that any food, drug, device or cosmetic has been adulterated or is being prepared, packed, or held under conditions whereby it may become adulterated or rendered injurious to health.
The Form 483 is a list of observations that the audit inspectors find objectionable at the plant and hand it out to the management. After receiving this, the company needs to reply in writing, within 15 days, giving a detailed plan of corrective action.
27-Nov-14 (Thursday): Here is what the company had to say:
The company had received some inspectional observations from the USFDA after the visit to their API manufacturing facility in Srikakulam district of Andhra Pradesh. The Company is committed to respond to the agency within stipulated timelines with their remedial plans and start implementing the necessary measures immediately.
At this stage, it has no implication on any activity at the plant i.e. unlikely to affect the production of the company. Hence, these are not expected to be material to the Company’s operations or consolidated results.
29-Jan-15 (Thursday): Earnings Call
With the release of the Q3-2015 Earnings’ call, the company confirmed that it has responded to the USFDA with a corrective action plan over a month ago in response to the Form 483, which it had received from the USFDA for the Srikakulam plant.
Simultaneously, the FDA continued their inspection of the facilities at Srikakulam, Miryalaguda, and Duvvada.
Month of February, 2015 : The US FDA adds 2 other facilities in its inspection list – the API manufacturing Miryalaguda, Telangana, as well as the Oncology Formulation manufacturing facility at Duvvada, Visakhapatnam, Andhra Pradesh in addition to the API facility at Srikakulam, Andhra Pradesh. With this, 3 of Dr. Reddy’s facilities come under scrutiny by the US Food and Drug Administration (FDA).
Here is a network of Dr. Reddy’s in India

5-Nov-15 (Thursday): 8 months later, USFDA issues a warning letter
USFDA issued a warning letter relating to Dr. Reddy’s API manufacturing facilities at Srikakulam, Andhra Pradesh and Miryalaguda, Telangana, as well as the Oncology Formulation manufacturing facility at Duvvada, Visakhapatnam, Andhra Pradesh. The action follows the earlier inspections of these sites by the agency in November 2014, January 2015 and February 2015 respectively.
Generally the warning letters are issued if the Form 483 observations are not confirmed at a later stage, followed by an import alert. This generally means that “The FDA has expressed disappointment towards their initial response to observations made In Form 483. It also means that no new applications will be approved until the initial observations are resolved or the audit concerns are addressed to the likes of the FDA.”
What does this mean? – The FDA Form 483 does not constitute a final Agency determination of whether any condition is in violation of the FD&C Act or any of its relevant regulations. The FDA Form 483 is considered, along with a written report called an Establishment Inspection Report, all evidence or documentation collected on-site, and any responses made by the company. The Agency considers all of this information and then determines what further action, if any, is appropriate to protect public health.
6-Nov-15 (Friday): Dr. Reddy’s confirms the receipt of the Warning Letter from the US FDA
Dr. Reddy’s CEO, G V Prasad commented, “We take quality and compliance matters seriously and stand by our commitment to fully comply with the cGMP quality standards across all our facilities. We will respond with a comprehensive plan to address these observations within the stipulated time-frame of 15 days. We will continue to actively engage with the agency to resolve these issues and we have also embarked on an initiate to revamp our quality systems and processed, as an organization-wide priority.”
The stock market also gets hold of this information and goes bonkers. The stock price dives from Rs. 4,283 to Rs. 3,635 in a single session.
9-Nov-15 (Monday): To calm market sentiments, Dr. Reddy’s held a Conference Call
Here is what was said:
Representatives of Dr. Reddy’s : Kedar Upadhye (Investor Relations), G. V. Prasad – Chief Executive Officer, Satish Reddy – Chairman, Saumen Chakraborty – Chief Financial Officer, and from the US – Abhijit Mukherjee – Chief Operating Officer.
At this stage we have just begun to compile the response on our comprehensive plan for the corrective and preventive actions for submission to the agency. This call is meant to give you a broad idea as to how we are approaching the matter. During the call we will disallow discussing of any specific observation made in the matter and we would also avoid any speculative comments on the next steps likely to be taken by the agency which would rightly be very inappropriate.
The letter mentions 3 sites – the API manufacturing facilities at Srikakulam, Andhra Pradesh and Miryalaguda, Telangana, as well as the Oncology Formulation manufacturing facility at Duvvada, Visakhapatnam, Andhra Pradesh.
We have been given 15 day’s time to respond and this matter is resolved the agency may withhold approval of any new application which involve these sites. The issue cited in the letter are cGMP violations relating primarily to documentation practices and controls, laboratory testing procedures, incidents investigation practices and well as standard operating procedures.
At this time we feel confident in the safety and ethicacy of our products, however we plan to do a comprehensive assessment of any risks relating to quality of our products. And there is no directive to stop the manufacturing activity or shipment of any products from these sites.
As we respond to the agency, it is imperative for us to continue to strengthen our quality management systems and processes and enhance the infrastructure for training and development of our staff on the current cGMP practices.
We have instituted corrective actions to address the form 483 observations received earlier in each of these sites which formed part of the updates shared with the agency. This recent letter underscores the need for us to re-evaluate the work done in light of the observations raised and continue to implement CAPA (Corrective & Preventive Actions) fully and assess the impact of FDA observations on our products as well as enhance our overall quality management systems.
We would also need to perform additional detailed third party assessment of our quality systems and evaluate our goal for global manufacturing operations to ensure compliance with cGMP regulations.
While this work is on, we will parallely attempt to de-risk supplies by transferring select products to alternate sites. We have already taken steps to implement such transfers and there is a dedicated team actively focussed on this activity. However, considering the amount of work involved in such transfers and capacity constraints, we should be prioritizing only select products for this process.
Clearly we are not happy with the fact that we could not resolve the form 483 observations to the agency’s satisfaction. We have always taken quality and compliance matters seriously and we will continue to remain focussed on the remedial measures. We have embarked on an initiative to revamp our quality systems and processes as a top organizational priority. We will obviously not compromise on making any required investments in terms of investments, training, consultancy as well as other areas as maybe required to bring us back into compliance.
Our Take
Though the conference call did assure the markets, the core reason why the stock fell was that the remediation plans submitted by Dr. Reddy’s was not to the liking of the USFDA. They apparently need a more comprehensive approach.
Here are some excerpts from the Q&A’s:
Neha Manpuria from J.P. Morgan
Q: You also mentioned that you were obviously taking corrective actions to address the observations raised in Form 483 which haven’t been satisfactory as per the USFDA and hence the warning letter. How would you go about identifying as to what additionally needs to be done to address what’s required in the warning letter?
A: The FDA has also given us some areas they would like to see specifically. It is in the areas of third party evaluations of product quality and third party verification of some of the actions we have taken and things like that. We have a fairly good idea of what their expectations are from the warning letter itself as well as from the observations had in before in the warning letter are a subset of the Form 483 observations which clearly carry a few themes and we understand those themes and we are using expert advice to help us in und
erstanding what else we should be doing other than the actions we have taken so far to meet FDA’s expectations. We are seriously working on that and in the next 2 weeks we will come out with a comprehensive plan to be submitted to the FDA.
Prakash Agarwal of Axis Capital
Q: Just trying to understand this procedure better; in October you got this OAI and it results in a warning letter. Is there a typical procedure that Form 483 and then the OAI and then the warning letter?
A: The fact that it’s an OAI is not very clear when the abbreviations are given. The FDA maintains a website where they indicate whether it’s OAI or potential OAI or VAI. I think if you see the recent change in the FDA’s way of working, we are also learning the process as we go forward. We have been giving regular updates and responses but we didn’t have any feedback clearly from the FDA on this matter. So beyond this we are not clear as to how this system works within the FDA.
Girish Bakhru of HSBC
Q: When you say you will have to take the measures across the network. Is there any impact onfacilities outside these three that you need to be cautious on?
A: Absolutely. Any corrective measure that we take has to be implemented at all our sites and we have been rolling out a numbers of enhancements across the network in the last several months and we still have to work out whether we need have third party verifications for the affected sites or across the network – that is something that we are still deliberating.Bachupally recently had its site inspection.
Here is the verbatim transcript of Saumen Chakraborty’s interview with Nigel D’Souza, Reema Tendulkar and Ekta Batra on CNBC-TV18. (US FDA Warning: Have 15 days to respond)
Here is the transcript of Surya Patra’s interview with Ekta Batra and Anuj Singhal on CNBC-TV18. (Pharma Companies facing pressue)
Now let’s get into the details of what happened : Part 2 – The Temporary Restraining Order
Though we would assume that the fire has been douzed, Dr. Reddy’s received a Temporary Restraining Order for the sale, delivery, transfer or other disposition of its generic esomeprazole product in the US market.

Our Take
This could be because Astra Zeneca – a British-Swedish multinational pharmaceutical and biologics company headquartered in London supposedly has a purple logo. If that’s the case, the fix should be quick and simple. But it is difficult to judge the matter at this moment, we will wait for the court hearing scheduled 2 days later on the 12-Nov-15.
Surprisingly, they’re friends too. Dr Reddy’s Laboratories Limited and AstraZeneca Pharma India Limited also have a distribution agreement for saxagliptin and its fixed dose combination with metformin, in Type 2 Diabetes.
Dr. Reddy’s is a good company and has fallen on the wrong side of the FDA. If the violations are addressed soon, it will probably overcome these issues and go back to its recent highs; but if it doesn’t, the stock will fall because they won’t be able to export to the US through these plants. The company’s response and the US FDA clearance (or lack of it) will determine its immediate future.
![]()
Disclaimer
Nothing in this newsletter is financial advice and should not be construed as such. Please do not take trading decisions based solely on the matter above; if you do, it is entirely at your own risk without any liability to Capital Mind. This is educational or informational matter only, and is provided as an opinion.
Disclosure: The authors at Capital Mind have positions in the market and some of them may support or contradict the material given above, or may involve a direction derived from independent analysis.

[/level-capmind-pro]